Cell Biology Antibodies 13
Anti-BBS7 Antibody (CAB17718)
(No reviews yet)
Write a Review
")
- SKU:
- CAB17718
")
Description
| Antibody Name: | Anti-BBS7 Antibody |
| Antibody SKU: | CAB17718 |
| Antibody Size: | 20uL, 50uL, 100uL |
| Application: | WB IHC IF |
| Reactivity: | Human, Mouse, Rat |
| Host Species: | Rabbit |
| Immunogen: | Recombinant fusion protein containing a sequence corresponding to amino acids 1-270 of human BBS7 (NP_060660.2). |
| Application: | WB IHC IF |
| Recommended Dilution: | WB 1:500 - 1:2000 IHC 1:50 - 1:100 IF 1:50 - 1:200 |
| Reactivity: | Human, Mouse, Rat |
| Positive Samples: | Rat testis, Rat lung |
| Immunogen: | Recombinant fusion protein containing a sequence corresponding to amino acids 1-270 of human BBS7 (NP_060660.2). |
| Purification Method: | Affinity purification |
| Storage Buffer: | Store at -20°C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Isotype: | IgG |
| Sequence: | MDLI LNRM DYLQ VGVT SQKT MKLI PASR HRAT QKVV IGDH DGVV MCFG MKKG EAAA VFKT LPGP KIAR LELG GVIN TPQE KIFI AAAS EIRG FTKR GKQF LSFE TNLT ESIK AMHI SGSD LFLS ASYI YNHY CDCK DQHY YLSG DKIN DVIC LPVE RLSR ITPV LACQ DRVL RVLQ GSDV MYAV EVPG PPTV LALH NGNG GDSG EDLL FGTS DGKL ALIQ ITTS KPVR KWEI QNEK KRGG ILCI DSFD IVGD GVKD LLVG RDDG MVEV YS |
| Gene ID: | 55212 |
| Uniprot: | Q8IWZ6 |
| Cellular Location: | |
| Calculated MW: | 75kDa/80kDa |
| Observed MW: | 80kDa |
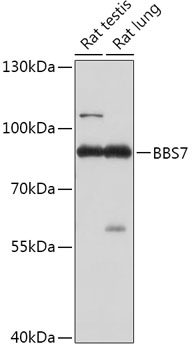 | Western blot - BBS7 Polyclonal Antibody (CAB17718) |
| Synonyms: | BBS2L1, BBS7 |
| Background: | This gene encodes one of eight proteins that form the BBSome complex containing BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9 and BBIP10. The BBSome complex is believed to recruit Rab8(GTP) to the primary cilium and promote ciliogenesis. The BBSome complex assembly is mediated by a complex composed of three chaperonin-like BBS proteins (BBS6, BBS10, and BBS12) and CCT/TRiC family chaperonins. Mutations in this gene are implicated in Bardet-Biedl syndrome, a genetic disorder whose symptoms include obesity, retinal degeneration, polydactyly and nephropathy; however, mutations in this gene and the BBS8 gene are thought to play a minor role and mutations in chaperonin-like BBS genes are found to be a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. Two transcript variants encoding distinct isoforms have been identified for this gene.[provided by RefSeq, Oct 2014] |
| UniProt Protein Function: | BBS7: The BBSome complex is required for ciliogenesis but is dispensable for centriolar satellite function. This ciliogenic function is mediated in part by the Rab8 GDP/GTP exchange factor, which localizes to the basal body and contacts the BBSome. Rab8(GTP) enters the primary cilium and promotes extension of the ciliary membrane. Firstly the BBSome associates with the ciliary membrane and binds to RAB3IP/Rabin8, the guanosyl exchange factor (GEF) for Rab8 and then the Rab8-GTP localizes to the cilium and promotes docking and fusion of carrier vesicles to the base of the ciliary membrane. Ciliary dysfunction leads to a broad spectrum of disorders, collectively termed ciliopathies. Overlapping clinical features include retinal degeneration, renal cystic disease, skeletal abnormalities, fibrosis of various organ, and a complex range of anatomical and functional defects of the central and peripheral nervous system. The ciliopathy range of diseases includes Meckel-Gruber syndrome, Bardet-Biedl syndrome, Joubert syndrome, nephronophtisis, Senior-Loken syndrome, and Jeune asphyxiating thoracic dystrophy among others. Single-locus allelism is insufficient to explain the variable penetrance and expressivity of such disorders, leading to the suggestion that variations across multiple sites of the ciliary proteome, including BBS7, influence the clinical outcome. Defects in BBS7 are a cause of Bardet-Biedl syndrome type 7 (BBS7). Bardet-Biedl syndrome (BBS) is a genetically heterogeneous disorder characterized by usually severe pigmentary retinopathy, early onset obesity, polydactyly, hypogenitalism, renal malformation and mental retardation. Secondary features include diabetes mellitus, hypertension and congenital heart disease. A relatively high incidence of BBS is found in the mixed Arab populations of Kuwait and in Bedouin tribes throughout the Middle East, most likely due to the high rate of consaguinity in these populations and a founder effect. Inheritance is autosomal recessive, but three mutated alleles (two at one locus, and a third at a second locus) may be required for disease manifestation in some cases (triallelic inheritance). 2 isoforms of the human protein are produced by alternative splicing. |
| UniProt Protein Details: | Chromosomal Location of Human Ortholog: 4q27 Cellular Component: axoneme; centrosome; cytosol; membrane Molecular Function:protein binding Biological Process: cilium biogenesis; determination of left/right symmetry; digestive tract morphogenesis; fat cell differentiation; heart looping; intracellular transport; melanosome transport; pigment granule aggregation in cell center; positive regulation of proteasomal ubiquitin-dependent protein catabolic process; protein localization; regulation of transcription from RNA polymerase II promoter Disease: Bardet-biedl Syndrome 7 |
| NCBI Summary: | This gene encodes one of eight proteins that form the BBSome complex containing BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9 and BBIP10. The BBSome complex is believed to recruit Rab8(GTP) to the primary cilium and promote ciliogenesis. The BBSome complex assembly is mediated by a complex composed of three chaperonin-like BBS proteins (BBS6, BBS10, and BBS12) and CCT/TRiC family chaperonins. Mutations in this gene are implicated in Bardet-Biedl syndrome, a genetic disorder whose symptoms include obesity, retinal degeneration, polydactyly and nephropathy; however, mutations in this gene and the BBS8 gene are thought to play a minor role and mutations in chaperonin-like BBS genes are found to be a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. Two transcript variants encoding distinct isoforms have been identified for this gene.[provided by RefSeq, Oct 2014] |
| UniProt Code: | Q8IWZ6 |
| NCBI GenInfo Identifier: | 90110978 |
| NCBI Gene ID: | 55212 |
| NCBI Accession: | Q8IWZ6.2 |
| UniProt Secondary Accession: | Q8IWZ6,Q4W5P8, Q8N581, Q9NVI4, |
| UniProt Related Accession: | Q8IWZ6 |
| Molecular Weight: | 75,446 Da |
| NCBI Full Name: | Bardet-Biedl syndrome 7 protein |
| NCBI Synonym Full Names: | Bardet-Biedl syndrome 7 |
| NCBI Official Symbol: | BBS7 |
| NCBI Official Synonym Symbols: | BBS2L1 |
| NCBI Protein Information: | Bardet-Biedl syndrome 7 protein |
| UniProt Protein Name: | Bardet-Biedl syndrome 7 protein |
| UniProt Synonym Protein Names: | BBS2-like protein 1 |
| Protein Family: | Bardet-Biedl syndrome 7 protein |
| UniProt Gene Name: | BBS7 |
| UniProt Entry Name: | BBS7_HUMAN |
Additional Information
Product Type: |
Antibody |
Reactivity: |
Human |
Reactivity: |
Mouse |
Reactivity: |
Rat |
Host Species: |
Rabbit |
Isotype: |
IgG |
Antibody Type: |
Polyclonal Antibody |
Research Area: |
Cell Biology |
Related Products
 ELISA Kit")
Human anti-EPO antibody(anti-Erythropoietin antibody) ELISA Kit
system_update_altDatasheet - तकनीकी मैनुअलsystem_update_altMSDS - तकनीक
")

 ELISA Kit")
Human Anti-DSG3 antibody(anti-Desmoglein-3 antibody) ELISA Kit
system_update_altDatasheet - तकनीकी मैनुअलsystem_update_altMSDS - तकनीक
ELISA Kit")
Human anti-ASGPR antibody(anti asialoglyco protein receptor antibody)ELISA Kit
OverviewProduct Name:Human anti-ASGPR antibody(anti asialoglyco protein receptor antibody)ELISA KitProduct Code:AEFI00360Size:96TAlias:a