ABCC9 Antibody (PACO20627)
")
- SKU:
- PACO20627
")
Description
| Antibody Name: | ABCC9 Antibody (PACO20627) |
| Antibody SKU: | PACO20627 |
| Size: | 50ul |
| Host Species: | Rabbit |
| Tested Applications: | ELISA, WB, IHC |
| Recommended Dilutions: | ELISA:1:1000-1:2000, WB:1:200-1:1000, IHC:1:40-1:150 |
| Species Reactivity: | Human, Mouse, Rat |
| Immunogen: | Synthetic peptide of human ABCC9 |
| Form: | Liquid |
| Storage Buffer: | -20°C, pH7.4 PBS, 0.05% NaN3, 40% Glycerol |
| Purification Method: | Antigen affinity purification |
| Clonality: | Polyclonal |
| Isotype: | IgG |
| Conjugate: | Non-conjugated |
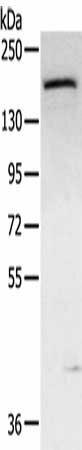 | Gel: 6%SDS-PAGE, Lysate: 40 µg, Lane: Mouse bladder tissue, Primary antibody: PACO20627(ABCC9 Antibody) at dilution 1/300, Secondary antibody: Goat anti rabbit IgG at 1/8000 dilution, Exposure time: 5 minutes. |
 | The image on the left is immunohistochemistry of paraffin-embedded Human liver cancer tissue using PACO20627(ABCC9 Antibody) at dilution 1/35, on the right is treated with synthetic peptide. (Original magnification: x200). |
| Background: | The protein encoded by this gene is a member of the superfamily of ATP-binding cassette (ABC) transporters. ABC proteins transport various molecules across extra- and intra-cellular membranes. ABC genes are divided into seven distinct subfamilies (ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, White). This protein is a member of the MRP subfamily which is involved in multi-drug resistance. This protein is thought to form ATP-sensitive potassium channels in cardiac, skeletal, and vascular and non-vascular smooth muscle. |
| Synonyms: | ATP-binding cassette, sub-family C (CFTR/MRP), member 9 |
| UniProt Protein Function: | ABCC9: Subunit of ATP-sensitive potassium channels (KATP). Can form cardiac and smooth muscle-type KATP channels with KCNJ11. KCNJ11 forms the channel pore while ABCC9 is required for activation and regulation. Defects in ABCC9 are the cause of cardiomyopathy dilated type 1O (CMD1O); also known as dilated cardiomyopathy with ventricular tachycardia. Dilated cardiomyopathy is a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. Defects in ABCC9 are the cause of familial atrial fibrillation type 12 (ATFB12). ATFB12 is a familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. Defects in ABCC9 are the cause of hypertrichotic osteochondrodysplasia (HTOCD). A rare disorder characterized by congenital hypertrichosis, neonatal macrosomia, a distinct osteochondrodysplasia, and cardiomegaly. The hypertrichosis leads to thick scalp hair, which extends onto the forehead, and a general increase in body hair. In addition, macrocephaly and coarse facial features, including a broad nasal bridge, epicanthal folds, a wide mouth, and full lips, can be suggestive of a storage disorder. About half of affected individuals are macrosomic and edematous at birth, whereas in childhood they usually have a muscular appearance with little subcutaneous fat. Thickened calvarium, narrow thorax, wide ribs, flattened or ovoid vertebral bodies, coxa valga, osteopenia, enlarged medullary canals, and metaphyseal widening of long bones have been reported. Cardiac manifestations such as patent ductus arteriosus, ventricular hypertrophy, pulmonary hypertension, and pericardial effusions are present in approximately 80% of cases. Motor development is usually delayed due to hypotonia. Most patients have a mild speech delay, and a small percentage have learning difficulties or intellectual disability. Belongs to the ABC transporter superfamily. ABCC family. Conjugate transporter (TC 3.A.1.208) subfamily. 2 isoforms of the human protein are produced by alternative splicing.Protein type: Membrane protein, integral; Membrane protein, multi-pass; Transporter; Transporter, ABC family; Channel, potassiumChromosomal Location of Human Ortholog: 12p12.1Cellular Component: ATP-sensitive potassium channel complex; plasma membrane; voltage-gated potassium channel complexMolecular Function: anion transmembrane-transporting ATPase activity; potassium channel regulator activity; sulfonylurea receptor activity; transporter activityBiological Process: defense response to virus; potassium ion import; potassium ion transport; transmembrane transportDisease: Atrial Fibrillation, Familial, 12; Cantu Syndrome; Cardiomyopathy, Dilated, 1o |
| UniProt Protein Details: | |
| NCBI Summary: | The protein encoded by this gene is a member of the superfamily of ATP-binding cassette (ABC) transporters. ABC proteins transport various molecules across extra- and intra-cellular membranes. ABC genes are divided into seven distinct subfamilies (ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, White). This protein is a member of the MRP subfamily which is involved in multi-drug resistance. This protein is thought to form ATP-sensitive potassium channels in cardiac, skeletal, and vascular and non-vascular smooth muscle. Protein structure suggests a role as the drug-binding channel-modulating subunit of the extra-pancreatic ATP-sensitive potassium channels. Mutations in this gene are associated with cardiomyopathy dilated type 1O. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Apr 2011] |
| UniProt Code: | O60706 |
| NCBI GenInfo Identifier: | 215273925 |
| NCBI Gene ID: | 10060 |
| NCBI Accession: | O60706.2 |
| UniProt Secondary Accession: | O60706,O60707 |
| UniProt Related Accession: | O60706 |
| Molecular Weight: | 174,425 Da |
| NCBI Full Name: | ATP-binding cassette sub-family C member 9 |
| NCBI Synonym Full Names: | ATP binding cassette subfamily C member 9 |
| NCBI Official Symbol: | ABCC9 |
| NCBI Official Synonym Symbols: | SUR2; ABC37; CANTU; CMD1O; ATFB12 |
| NCBI Protein Information: | ATP-binding cassette sub-family C member 9 |
| UniProt Protein Name: | ATP-binding cassette sub-family C member 9 |
| UniProt Synonym Protein Names: | Sulfonylurea receptor 2 |
| Protein Family: | ABC transporter C family |
| UniProt Gene Name: | ABCC9 |
| UniProt Entry Name: | ABCC9_HUMAN |
Additional Information
Product Type: |
Antibody |
Reactivity: |
Human |
Reactivity: |
Mouse |
Reactivity: |
Rat |
Host Species: |
Rabbit |
Isotype: |
IgG |
Applications: |
ELISA |
Applications: |
WB |
Applications: |
IHC |
Antibody Type: |
Polyclonal Antibody |
Conjugation: |
Unconjugated |
Related Products
")
")
 ELISA Kit")
 ELISA Kit")
Human TP53 antibody(p53 antibody) ELISA Kit
system_update_altDatasheet - तकनीकी मैनुअलsystem_update_altMSDS - तकनीक

CYP2A13 Antibody
Overview Product Name:CYP2A13 AntibodyProduct Code:PACO64657Size:50µLTarget Names:CYP2A13Species Reactivity:Human, Mouse