Epigenetics & Nuclear Signaling Antibodies 4
Anti-PMS2 Antibody (CAB6947)
(No reviews yet)
Write a Review
")
- SKU:
- CAB6947
")
Description
| Antibody Name: | Anti-PMS2 Antibody |
| Antibody SKU: | CAB6947 |
| Antibody Size: | 20uL, 50uL, 100uL |
| Application: | WB IF |
| Reactivity: | Human, Mouse, Rat |
| Host Species: | Rabbit |
| Immunogen: | Recombinant fusion protein containing a sequence corresponding to amino acids 390-670 of human PMS2 (NP_000526.2). |
| Application: | WB IF |
| Recommended Dilution: | WB 1:500 - 1:2000 IF 1:50 - 1:100 |
| Reactivity: | Human, Mouse, Rat |
| Positive Samples: | Jurkat, A-431, HeLa, Mouse brain, Mouse thymus, Rat brain |
| Immunogen: | Recombinant fusion protein containing a sequence corresponding to amino acids 390-670 of human PMS2 (NP_000526.2). |
| Purification Method: | Affinity purification |
| Storage Buffer: | Store at -20'C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Isotype: | IgG |
| Sequence: | ADLE KPMV EKQD QSPS LRTG EEKK DVSI SRLR EAFS LRHT TENK PHSP KTPE PRRS PLGQ KRGM LSSS TSGA ISDK GVLR PQKE AVSS SHGP SDPT DRAE VEKD SGHG STSV DSEG FSIP DTGS HCSS EYAA SSPG DRGS QEHV DSQE KAPK TDDS FSDV DCHS NQED TGCK FRVL PQPT NLAT PNTK RFKK EEIL SSSD ICQK LVNT QDMS ASQV DVAV KINK KVVP LDFS MSSL AKRI KQLH HEAQ QSEG EQNY RKFR AKIC PGEN QAAE DELR KEIS K |
| Gene ID: | 5395 |
| Uniprot: | P54278 |
| Cellular Location: | Nucleus |
| Calculated MW: | 20kDa/51kDa/62kDa/95kDa |
| Observed MW: | 96kDa, 115kDa |
| Synonyms: | PMS2, HNPCC4, MLH4, PMS2CL, PMSL2 |
| Background: | The protein encoded by this gene is a key component of the mismatch repair system that functions to correct DNA mismatches and small insertions and deletions that can occur during DNA replication and homologous recombination. This protein forms heterodimers with the gene product of the mutL homolog 1 (MLH1) gene to form the MutL-alpha heterodimer. The MutL-alpha heterodimer possesses an endonucleolytic activity that is activated following recognition of mismatches and insertion/deletion loops by the MutS-alpha and MutS-beta heterodimers, and is necessary for removal of the mismatched DNA. There is a DQHA(X)2E(X)4E motif found at the C-terminus of the protein encoded by this gene that forms part of the active site of the nuclease. Mutations in this gene have been associated with hereditary nonpolyposis colorectal cancer (HNPCC; also known as Lynch syndrome) and Turcot syndrome. |
| UniProt Protein Function: | PMS2: Component of the post-replicative DNA mismatch repair system (MMR). Heterodimerizes with MLH1 to form MutL alpha. DNA repair is initiated by MutS alpha (MSH2-MSH6) or MutS beta (MSH2- MSH6) binding to a dsDNA mismatch, then MutL alpha is recruited to the heteroduplex. Assembly of the MutL-MutS-heteroduplex ternary complex in presence of RFC and PCNA is sufficient to activate endonuclease activity of PMS2. It introduces single-strand breaks near the mismatch and thus generates new entry points for the exonuclease EXO1 to degrade the strand containing the mismatch. DNA methylation would prevent cleavage and therefore assure that only the newly mutated DNA strand is going to be corrected. MulL alpha (MLH1-PMS2) interacts physically with the clamp loader subunits of DNA polymerase III, suggesting that it may play a role to recruit the DNA polymerase III to the site of the MMR. Also implicated in DNA damage signaling, a process which induces cell cycle arrest and can lead to apoptosis in case of major DNA damages. Defects in PMS2 are the cause of hereditary non-polyposis colorectal cancer type 4 (HNPCC4). Mutations in more than one gene locus can be involved alone or in combination in the production of the HNPCC phenotype (also called Lynch syndrome). Most families with clinically recognized HNPCC have mutations in either MLH1 or MSH2 genes. HNPCC is an autosomal, dominantly inherited disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early onset colorectal carcinoma (CRC) and extra-colonic cancers of the gastrointestinal, urological and female reproductive tracts. HNPCC is reported to be the most common form of inherited colorectal cancer in the Western world, and accounts for 15% of all colon cancers. Cancers in HNPCC originate within benign neoplastic polyps termed adenomas. Clinically, HNPCC is often divided into two subgroups. Type I: hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II: patients have an increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical HNPCC is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected HNPCC' or 'incomplete HNPCC' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. Defects in PMS2 are a cause of mismatch repair cancer syndrome (MMRCS); also known as Turcot syndrome or brain tumor-polyposis syndrome 1 (BTPS1). MMRCS is an autosomal dominant disorder characterized by malignant tumors of the brain associated with multiple colorectal adenomas. Skin features include sebaceous cysts, hyperpigmented and cafe au lait spots. Belongs to the DNA mismatch repair MutL/HexB family. 4 isoforms of the human protein are produced by alternative splicing. |
| UniProt Protein Details: | Protein type:Tumor suppressor; Hydrolase; EC 3.1.-.-; DNA repair, damage; Cell cycle regulation Chromosomal Location of Human Ortholog: 7p22.2 Cellular Component: nucleoplasm; MutLalpha complex; microtubule cytoskeleton; cytoplasm; nucleus Molecular Function:protein binding; DNA binding; single base insertion or deletion binding; endonuclease activity; ATPase activity; MutSalpha complex binding; ATP binding; single-stranded DNA binding Biological Process: response to drug; mismatch repair; somatic hypermutation of immunoglobulin genes; somatic recombination of immunoglobulin gene segments; DNA repair Disease: Colorectal Cancer, Hereditary Nonpolyposis, Type 4; Mismatch Repair Cancer Syndrome |
| NCBI Summary: | This gene is one of the PMS2 gene family members found in clusters on chromosome 7. The product of this gene is involved in DNA mismatch repair. It forms a heterodimer with MLH1 and this complex interacts with other complexes bound to mismatched bases. Mutations in this gene are associated with hereditary nonpolyposis colorectal cancer, Turcot syndrome, and are a cause of supratentorial primitive neuroectodermal tumors. Alternatively spliced transcript variants have been observed for this gene. [provided by RefSeq, Jul 2008] |
| UniProt Code: | P54278 |
| NCBI GenInfo Identifier: | 317373266 |
| NCBI Gene ID: | 5395 |
| NCBI Accession: | P54278.2 |
| UniProt Secondary Accession: | P54278,Q52LH6, Q5FBW9, Q5FBX1, Q5FBX2, Q75MR2, B2R610 |
| UniProt Related Accession: | P54278 |
| Molecular Weight: | 862 |
| NCBI Full Name: | Mismatch repair endonuclease PMS2 |
| NCBI Synonym Full Names: | PMS2 postmeiotic segregation increased 2 (S. cerevisiae) |
| NCBI Official Symbol: | PMS2 |
| NCBI Official Synonym Symbols: | PMSL2; HNPCC4; PMS2CL |
| NCBI Protein Information: | mismatch repair endonuclease PMS2; H_DJ0042M02.9; PMS1 protein homolog 2; DNA mismatch repair protein PMS2 |
| UniProt Protein Name: | Mismatch repair endonuclease PMS2 |
| UniProt Synonym Protein Names: | DNA mismatch repair protein PMS2; PMS1 protein homolog 2 |
| Protein Family: | Mismatch repair endonuclease |
| UniProt Gene Name: | PMS2 |
| UniProt Entry Name: | PMS2_HUMAN |
 | Western blot analysis of extracts of various cell lines, using PMS2 antibody (CAB6947) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (RM00020). Exposure time: 90s. |
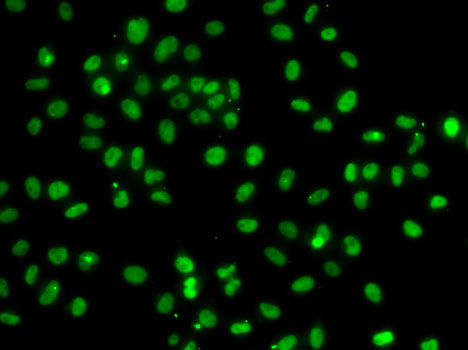 | Immunofluorescence analysis of A-549 cells using PMS2 antibody (CAB6947). |
View AllClose
Additional Information
Product Type: |
Antibody |
Reactivity: |
Human |
Reactivity: |
Mouse |
Reactivity: |
Rat |
Host Species: |
Rabbit |
Isotype: |
IgG |
Antibody Type: |
Polyclonal Antibody |
Research Area: |
Epigenetics and Nuclear Signaling |
Related Products
")
Anti-PMS2 Antibody (CAB4577)
Epigenetics & Nuclear Signaling Antibodies 5
OverviewProduct Name:PMS2 Rabbit mAbProduct Code:CAB4577Size:20uL, 50uL, 100uLSynonyms:HNPCC4, MLH4, PMS2CL, PMSL2Appl
")
![Anti-PMS2 Antibody (CAB19928)[KO Validated]](https://cdn11.bigcommerce.com/s-rd6ounxcu2/images/stencil/590x590/products/56096/61278/anti-pms2-antibody-cab19928ko-validated__50397__40230.1706533757.jpg?c=1 "Anti-PMS2 Antibody (CAB19928)[KO Validated]")
![Anti-PMS2 Antibody (CAB19927)[KO Validated]](https://cdn11.bigcommerce.com/s-rd6ounxcu2/images/stencil/590x590/products/56095/61277/anti-pms2-antibody-cab19927ko-validated__28351__23305.1706533757.jpg?c=1 "Anti-PMS2 Antibody (CAB19927)[KO Validated]")

PMS2 Monoclonal Antibody
OverviewProduct Name:PMS2 Monoclonal AntibodyProduct Code:CAB22350Reactivity:HumanApplications:Western blotting