Cell Biology Antibodies 4
Anti-IDUA Antibody (CAB13779)
(No reviews yet)
Write a Review
")
- SKU:
- CAB13779
")
Description
| Antibody Name: | Anti-IDUA Antibody |
| Antibody SKU: | CAB13779 |
| Antibody Size: | 20uL, 50uL, 100uL |
| Application: | WB |
| Reactivity: | Mouse |
| Host Species: | Rabbit |
| Immunogen: | Recombinant fusion protein containing a sequence corresponding to amino acids 354-653 of human IDUA (NP_000194.2). |
| Application: | WB |
| Recommended Dilution: | WB 1:500 - 1:2000 |
| Reactivity: | Mouse |
| Positive Samples: | Mouse brain |
| Immunogen: | Recombinant fusion protein containing a sequence corresponding to amino acids 354-653 of human IDUA (NP_000194.2). |
| Purification Method: | Affinity purification |
| Storage Buffer: | Store at -20'C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Isotype: | IgG |
| Sequence: | SYHP HPFA QRTL TARF QVNN TRPP HVQL LRKP VLTA MGLL ALLD EEQL WAEV SQAG TVLD SNHT VGVL ASAH RPQG PADA WRAA VLIY ASDD TRAH PNRS VAVT LRLR GVPP GPGL VYVT RYLD NGLC SPDG EWRR LGRP VFPT AEQF RRMR AAED PVAA APRP LPAG GRLT LRPA LRLP SLLL VHVC ARPE KPPG QVTR LRAL PLTQ GQLV LVWS DEHV GSKC LWTY EIQF SQDG KAYT PVSR KPST FNLF VFSP DTGA VSGS YRVR ALDY WARP GPFS DPVP YLEV PVPR GPPS PGNP |
| Gene ID: | 3425 |
| Uniprot: | P35475 |
| Cellular Location: | Lysosome |
| Calculated MW: | 72kDa/73kDa |
| Observed MW: | 65kDa |
| Synonyms: | IDUA, IDA, MPS1 |
| Background: | This gene encodes an enzyme that hydrolyzes the terminal alpha-L-iduronic acid residues of two glycosaminoglycans, dermatan sulfate and heparan sulfate. This hydrolysis is required for the lysosomal degradation of these glycosaminoglycans. Mutations in this gene that result in enzymatic deficiency lead to the autosomal recessive disease mucopolysaccharidosis type I (MPS I). |
| UniProt Protein Function: | IDUA: Defects in IDUA are the cause of mucopolysaccharidosis type 1H (MPS1H); also known as Hurler syndrome. MPS1H is a severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe mental retardation. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. Defects in IDUA are the cause of mucopolysaccharidosis type 1H/S (MPS1H/S); also known as Hurler-Scheie syndrome. MPS1H/S is a form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. Defects in IDUA are the cause of mucopolysaccharidosis type 1S (MPS1S); also known as Scheie syndrome. MPS1S is a mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. Belongs to the glycosyl hydrolase 39 family. |
| UniProt Protein Details: | Protein type:EC 3.2.1.76; Hydrolase; Glycan Metabolism - glycosaminoglycan degradation Chromosomal Location of Human Ortholog: 4p16.3 Cellular Component: lysosomal lumen Molecular Function:L-iduronidase activity; receptor binding Biological Process: carbohydrate metabolic process; cell morphogenesis; chemical homeostasis; chondroitin sulfate catabolic process; chondroitin sulfate metabolic process; dermatan sulfate catabolic process; disaccharide metabolic process; glycosaminoglycan catabolic process; glycosaminoglycan metabolic process; limb morphogenesis; lysosome organization and biogenesis; skeletal morphogenesis Disease: Hurler Syndrome; Hurler-scheie Syndrome; Scheie Syndrome |
| NCBI Summary: | This gene encodes an enzyme that hydrolyzes the terminal alpha-L-iduronic acid residues of two glycosaminoglycans, dermatan sulfate and heparan sulfate. This hydrolysis is required for the lysosomal degradation of these glycosaminoglycans. Mutations in this gene that result in enzymatic deficiency lead to the autosomal recessive disease mucopolysaccharidosis type I (MPS I). [provided by RefSeq, Jul 2008] |
| UniProt Code: | P35475 |
| NCBI GenInfo Identifier: | 110611239 |
| NCBI Gene ID: | 3425 |
| NCBI Accession: | NP_000194.2 |
| UniProt Secondary Accession: | P35475,B3KWK6, |
| UniProt Related Accession: | P35475 |
| Molecular Weight: | 71.9 kDa |
| NCBI Full Name: | alpha-L-iduronidase |
| NCBI Synonym Full Names: | iduronidase, alpha-L- |
| NCBI Official Symbol: | IDUA |
| NCBI Official Synonym Symbols: | IDA; MPS1 |
| NCBI Protein Information: | alpha-L-iduronidase |
| UniProt Protein Name: | Alpha-L-iduronidase |
| Protein Family: | Alpha-L-iduronidase |
| UniProt Gene Name: | IDUA |
| UniProt Entry Name: | IDUA_HUMAN |
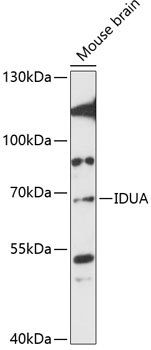 | Western blot analysis of extracts of mouse brain, using IDUA antibody (CAB13779) at 1:3000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Enhanced Kit (RM00021). Exposure time: 90s. |
View AllClose
Additional Information
Product Type: |
Antibody |
Reactivity: |
Mouse |
Host Species: |
Rabbit |
Isotype: |
IgG |
Antibody Type: |
Polyclonal Antibody |
Research Area: |
Cell Biology |
Related Products
")
Human IDUA PharmaGenie ELISA Kit (SBRS0693)
system_update_altDatasheetHuman IDUA PharmaGenie ELISA Kit (SBRS0693)Overview
 ELISA Kit")
Human anti-EPO antibody(anti-Erythropoietin antibody) ELISA Kit
system_update_altDatasheet - तकनीकी मैनुअलsystem_update_altMSDS - तकनीक
")

