Cell Biology Antibodies 7
Anti-ATXN2 Antibody (CAB16666)
(No reviews yet)
Write a Review
")
- SKU:
- CAB16666
")
Description
| Antibody Name: | Anti-ATXN2 Antibody |
| Antibody SKU: | CAB16666 |
| Antibody Size: | 20uL, 50uL, 100uL |
| Application: | WB IHC IF |
| Reactivity: | Human, Mouse, Rat |
| Host Species: | Rabbit |
| Immunogen: | A synthetic peptide of human ATXN2 |
| Application: | WB IHC IF |
| Recommended Dilution: | WB 1:500 - 1:2000 IHC 1:50 - 1:200 IF 1:50 - 1:200 |
| Reactivity: | Human, Mouse, Rat |
| Positive Samples: | U-87MG, Mouse liver, Rat liver |
| Immunogen: | A synthetic peptide of human ATXN2 |
| Purification Method: | Affinity purification |
| Storage Buffer: | Store at -20'C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Isotype: | IgG |
| Sequence: | Email for sequence |
| Gene ID: | 6311 |
| Uniprot: | Q99700 |
| Cellular Location: | Cytoplasm |
| Calculated MW: | 27kDa/106kDa/109kDa/132kDa/140kDa |
| Observed MW: | 170kDa |
| Synonyms: | ATXN2, ATX2, SCA2, TNRC13, ataxin-2 |
| Background: | This gene belongs to a group of genes that is associated with microsatellite-expansion diseases, a class of neurological and neuromuscular disorders caused by expansion of short stretches of repetitive DNA. The protein encoded by this gene has two globular domains near the N-terminus, one of which contains a clathrin-mediated trans-Golgi signal and an endoplasmic reticulum exit signal. The encoded cytoplasmic protein localizes to the endoplasmic reticulum and plasma membrane, is involved in endocytosis, and modulates mTOR signals, modifying ribosomal translation and mitochondrial function. The N-terminal region of the protein contains a polyglutamine tract of 14-31 residues that can be expanded in the pathogenic state to 32-200 residues. Intermediate length expansions of this tract increase susceptibility to amyotrophic lateral sclerosis, while long expansions of this tract result in spinocerebellar ataxia-2, an autosomal-dominantly inherited, neurodegenerative disorder. Genome-wide association studies indicate that loss-of-function mutations in this gene may be associated with susceptibility to type I diabetes, obesity and hypertension. Alternative splicing results in multiple transcript variants. |
| UniProt Protein Function: | ataxin-2: Involved in EGFR trafficking, acting as negative regulator of endocytic EGFR internalization at the plasma membrane. Defects in ATXN2 are the cause of spinocerebellar ataxia type 2 (SCA2); also known as olivopontocerebellar atrophy II (OPCA II or OPCA2). Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to cerebellum degeneration with variable involvement of the brainstem and spinal cord. SCA2 belongs to the autosomal dominant cerebellar ataxias type I (ADCA I) which are characterized by cerebellar ataxia in combination with additional clinical features like optic atrophy, ophthalmoplegia, bulbar and extrapyramidal signs, peripheral neuropathy and dementia. SCA2 is characterized by hyporeflexia, myoclonus and action tremor and dopamine-responsive parkinsonism. SCA2 is caused by expansion of a CAG repeat resulting in about 36 to 52 repeats in some patients. Longer expansions result in earlier the expansion, onset of the disease. Defects in ATXN2 are a cause of susceptibility to amyotrophic lateral sclerosis type 13 (ALS13). It is a neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. Death usually occurs within 2 to 5 years. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. An increased risk for developing amyotrophic lateral sclerosis is seems to be conferred by CAG repeat intermediate expansions greater than 23 but below the threshold for developing spinocerebellar ataxia. Belongs to the ataxin-2 family. 4 isoforms of the human protein are produced by alternative splicing. |
| UniProt Protein Details: | Protein type:Translation; RNA-binding Chromosomal Location of Human Ortholog: 12q24.1 Cellular Component: cytoplasm; Golgi apparatus; membrane; nucleoplasm; perinuclear region of cytoplasm; polysome; ribonucleoprotein complex; stress granule; trans-Golgi network Molecular Function:epidermal growth factor receptor binding; protein binding; protein C-terminus binding Biological Process: cytoplasmic mRNA processing body assembly; negative regulation of receptor internalization; stress granule assembly Disease: Parkinson Disease, Late-onset; Spinocerebellar Ataxia 2 |
| NCBI Summary: | This gene belongs to a group of genes that is associated with microsatellite-expansion diseases, a class of neurological and neuromuscular disorders caused by expansion of short stretches of repetitive DNA. The protein encoded by this gene has two globular domains near the N-terminus, one of which contains a clathrin-mediated trans-Golgi signal and an endoplasmic reticulum exit signal. The protein is primarily localized to the Golgi apparatus, with deletion of the Golgi and endoplasmic reticulum signals resulting in abnormal subcellular localization. In addition, the N-terminal region contains a polyglutamine tract of 14-31 residues that can be expanded in the pathogenic state to 32-200 residues. Intermediate length expansions of this tract increase susceptibility to amyotrophic lateral sclerosis, while long expansions of this tract result in spinocerebellar ataxia-2, an autosomal-dominantly inherited, neurodegenerative disorder. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jul 2016] |
| UniProt Code: | Q99700 |
| NCBI GenInfo Identifier: | 215273941 |
| NCBI Gene ID: | 6311 |
| NCBI Accession: | Q99700.2 |
| UniProt Secondary Accession: | Q99700,Q24JQ7, Q6ZQZ7, Q99493, A6NLD4, |
| UniProt Related Accession: | Q99700 |
| Molecular Weight: | 109,037 Da |
| NCBI Full Name: | Ataxin-2 |
| NCBI Synonym Full Names: | ataxin 2 |
| NCBI Official Symbol: | ATXN2 |
| NCBI Official Synonym Symbols: | ATX2; SCA2; ASL13; TNRC13 |
| NCBI Protein Information: | ataxin-2 |
| UniProt Protein Name: | Ataxin-2 |
| UniProt Synonym Protein Names: | Spinocerebellar ataxia type 2 protein; Trinucleotide repeat-containing gene 13 protein |
| Protein Family: | Ataxin |
| UniProt Gene Name: | ATXN2 |
| UniProt Entry Name: | ATX2_HUMAN |
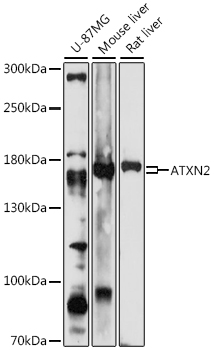 | Western blot analysis of extracts of various cell lines, using ATXN2 antibody (CAB16666) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Enhanced Kit (RM00021). Exposure time: 30s. |
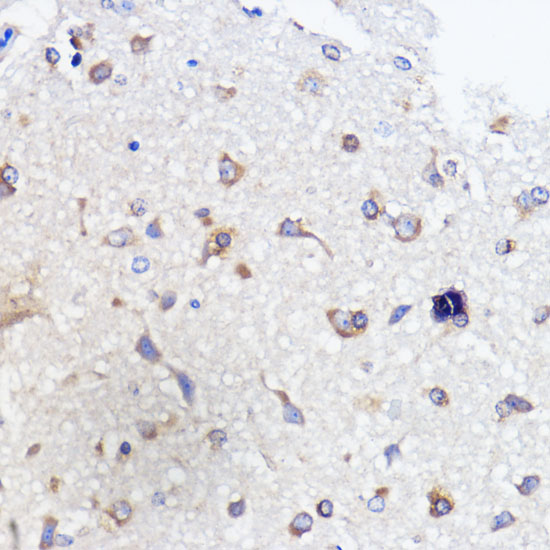 | Immunohistochemistry of paraffin-embedded rat brain using ATXN2 antibody (CAB16666) at dilution of 1:100 (40x lens). |
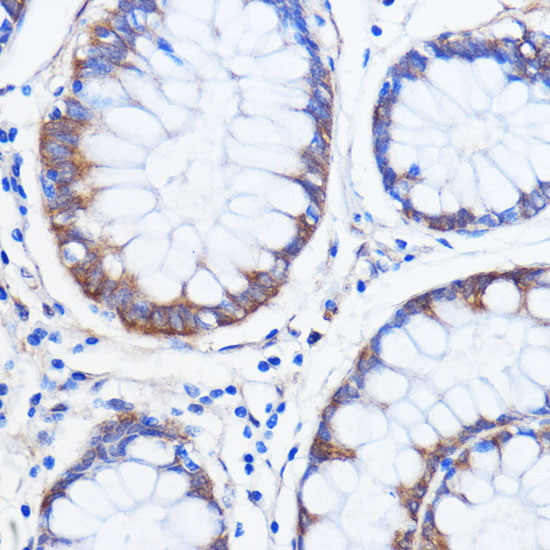 | Immunohistochemistry of paraffin-embedded human colon carcinoma using ATXN2 antibody (CAB16666) at dilution of 1:100 (40x lens). |
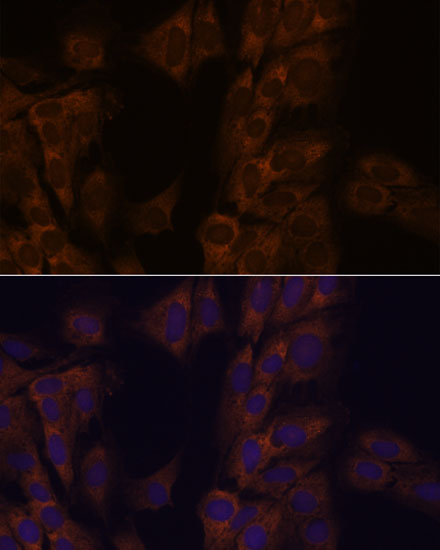 | Immunofluorescence analysis of C6 cells using ATXN2 antibody (CAB16666) at dilution of 1:100. Blue: DAPI for nuclear staining. |
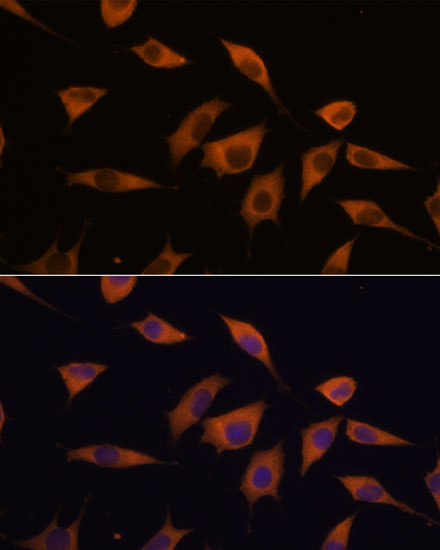 | Immunofluorescence analysis of L929 cells using ATXN2 antibody (CAB16666) at dilution of 1:100. Blue: DAPI for nuclear staining. |
View AllClose
Additional Information
Product Type: |
Antibody |
Reactivity: |
Human |
Reactivity: |
Mouse |
Reactivity: |
Rat |
Host Species: |
Rabbit |
Isotype: |
IgG |
Antibody Type: |
Polyclonal Antibody |
Research Area: |
Cell Biology |
Related Products
")
")
Human ATXN2 Recombinant Protein (GST tag)
OverviewProduct Name:Human ATXN2 Recombinant Protein (GST tag)Product Code:RPES5390Size:20µgSpecies:HumanExpres
 ELISA Kit (HUES01716)")
 ELISA Kit")
Rat ATXN2 (Ataxin 2) ELISA Kit
OverviewProduct Name:Rat ATXN2 (Ataxin 2) ELISA KitProduct Code:AEES00319Assay Type:SandwichFormat:96TAssay Tim
 ELISA Kit")
Human anti-EPO antibody(anti-Erythropoietin antibody) ELISA Kit
system_update_altDatasheet - तकनीकी मैनुअलsystem_update_altMSDS - तकनीक